Elucidating novel crystalline structures with electron and NMR crystallography
Author(s): Dr. Michael Frey, Analytical Instruments Product Manager, JEOL USA, Inc and Dr. Yusuke Nishiyama, Research Scientist, JEOL RESONANCE Inc
Determining the crystal structure of chemical compounds and biological molecules, by defining the precise spatial arrangements of their atoms in a crystalline state, is crucial to understanding their function and reactivity. In addition to crystalline structure, analyzing the hydrogen bonding strength between hydrogen and electronegative atoms is vital in applications such as medicine, engineering, materials science and pharmaceutical research. This is due to the influence of hydrogen bond strength on molecular function and inter- and intra-molecular packing stability. Elucidating the structure of molecular crystals at atomic resolution and defining the position of hydrogen atoms is critical to understanding their complex hydrogen-bonding networks.
In the pharmaceutical industry, low-molecular-weight orally administered drugs with a long shelf-life are required for treating adult diseases and hay fever etc. which require daily doses. Different crystal structures have different solubilities and stability, altering their therapeutic effects, so the structure of active pharmaceutical ingredients (APIs) must be controlled to ensure the stable efficacy of small molecule drugs.
Structure determination technology
Single-crystal X-ray diffraction (SCXRD) is a fundamental technique for determining the structure of large single crystals (>10-100 µm) and their hydrogen-bonding networks. However, smaller crystals rarely produce enough diffraction spots for structural elucidation and although powder X-ray diffraction (PXRD) may be used if a large amount of crystals is available, it cannot easily localize the hydrogen atoms and requires highly isomorphic microcrystalline samples with small lattices. PXRD also struggles to differentiate between atoms with similar atomic numbers, such as carbon, nitrogen and oxygen, making structure determination of organic molecules challenging.
Other approaches, such as infrared (IR) spectroscopy and solid-state nuclear magnetic resonance (SSNMR), are useful for detecting the presence of hydrogen bonds in nanocrystals and microcrystals, but whole-crystal structures often remain unobtainable. So, although SSNMR provides chemical shift values and information on the proximity between hydrogen atoms and internuclear distance measurements (1H-X [X=13C, 15N]), its combination with XRD and quantum computation is required to gain a comprehensive understanding of crystal structures and hydrogen-bonding networks of unknown compounds.
In this combined approach, termed NMR crystallography, XRD is used to determine the position of every atom in a crystal except hydrogen. Quantum computation then places the hydrogen atoms in rational positions to be verified using SSNMR, which directly measures nitrogen and carbon atoms and assures atomic assignments.
Advanced NMR crystallography
Despite the potential of XRD and SSNMR to drive crystal structure determination in a range of fields, the widespread use of NMR crystallography is limited by a number of challenges. For example, many pharmaceutical dosage forms such as tablets contain complex components, making XRD analysis difficult and therefore impacting the downstream SSNMR measurements. Electron diffraction (ED) – an observation mode of transmission electron microscopy (TEM) – goes some way to resolve this problem, by allowing the three-dimensional (3D) structures of nanocrystals to be solved. As an alternative diffraction method to XRD and neutron diffraction (ND), ED provides a method of observing scattering patterns by irradiating crystals with an electron beam and gives an electrostatic potential, whereas XRD gives an electron density distribution. Compared to X-rays and neutrons, the ED pattern can be obtained from very small crystals because electrons interact with material much stronger than X-rays and neutrons.
Shedding light on unknown pharmaceutical structures
One study conducted by researchers at Kyoto University, JEOL, RIKEN Spring-8 Center and the RIKEN-JEOL Collaboration Center used ED combined with SSNMR and quantum computation to confirm the microcrystal structure of the amino acid L-histidine (LH) as a proof of concept, before elucidating the previously unknown structure of cimetidine crystal form B (CB) [1]. Cimetidine (C10H16N6S) is a histamine H2 receptor antagonist that inhibits stomach acid production, used primarily in treating heartburn and peptic ulcers. It is known to take various crystal forms (A, B, C and D/Z) and the monohydrates (M1, M2 and M3). The needle-like structure of CB in its crystal form makes it challenging to produce a single crystal for XRD analysis, and it often crystallizes in a mixed state with crystal form C, limiting PXRD use.
The method first used ED to determine the nanocrystal structure of the organic samples, then employed SSNMR to solve the ambiguities originating from the misassignment of carbon, nitrogen and oxygen atoms, and ambiguous hydrogen atom identification. For example, ED of the pure orthorhombic LH resulted in only three of the nine hydrogen atoms being identified, and the positions of these were found to be inaccurate. Following refinement using SHELXL software [2], two candidate structures for LH were established – LH1(2) and LH3(4) – which differed only in terms of the hydrogen atom positions. It was therefore important to correctly determine the hydrogen atom locations, which was partially enabled by fast magic angle spinning (MAS) SSNMR to generate 1H, 14N and 15N signals. This gave rise to four possible structures of LH1-4.
Bond lengths were then determined by measuring 1H-X dipolar interactions using 2D inversely proton-detected cross-polarization with variable contact time (invCP-VC), and structures were finally refined with these bond lengths using SSNMR. To identify the correct structure, gauge-including projected augmented wave (GIPAW) calculations and SSNMR were combined in the NMR crystallography workflow. GIPAW optimizes a crystal structure by relaxing the atomic positions to local minima of the energy surface in addition to providing NMR parameters, such as isotropic chemical shifts and quadrupolar couplings. The results showed the LH1 structure to be the most energetically favorable, a finding which was ratified by root-mean-square deviation (RMSD) calculations.
This approach was then applied to determine the crystal structure of CB. Although 13C SSNMR spectra have been used to distinguish different crystal forms, B and C exhibit very similar chemical shifts and there are no distinct 13C chemical shifts corresponding to form C. As in LH structure determination, ED was used to solve the initial crystal structure of CB which is comprised of two different conformations: one with an extended conformation and the other with a bent conformation. Based on these conformations, four candidate models for CB were considered (Figure 1), and hydrogen positions were refined using the internuclear distances measured using SSNMR. GIPAW optimization showed that CB2 was the most energetically favorable, and the final validation was conducted by evaluating the RMSD values for the 1H, 13C and 15N chemical shifts, all of which indicated CB2 as the correct structure.
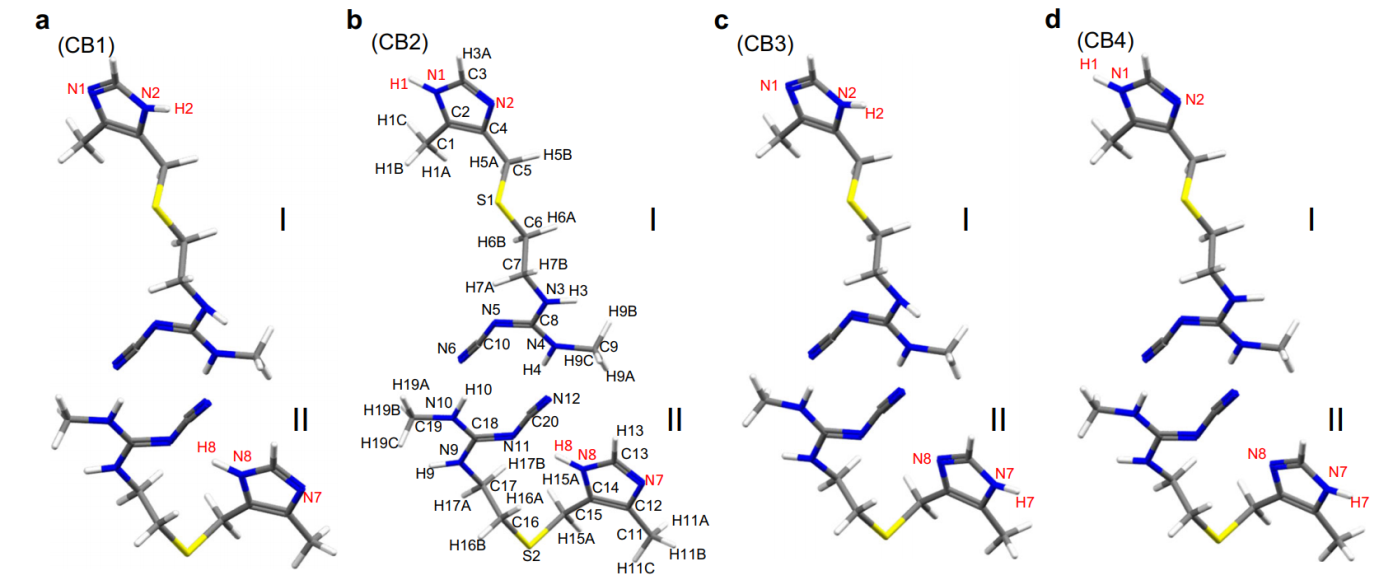
Figure 1: Four possible molecular structures of cimetidine form B (CB). The cimetidine form B (CB) structures of (a) CB1, (b) CB2, (c) CB3, and (d) CB4 depend on the protonation site of the imidazole ring. The atomic numbering scheme is shown. Each structure is composed of two conformations; I and II refers to an extended and a bent conformation, respectively. The yellow, blue, gray, and white atoms denote sulfur, nitrogen, carbon, and hydrogen atoms, respectively. Reproduced in accordance with Creative Commons Attribution 4.0 International License from reference [1].
Figure 2 shows the inter- and intra-molecular hydrogen-bonding network of CB2, highlighting the importance of the positions of hydrogen atoms on the conformation and crystal packing of pharmaceuticals.
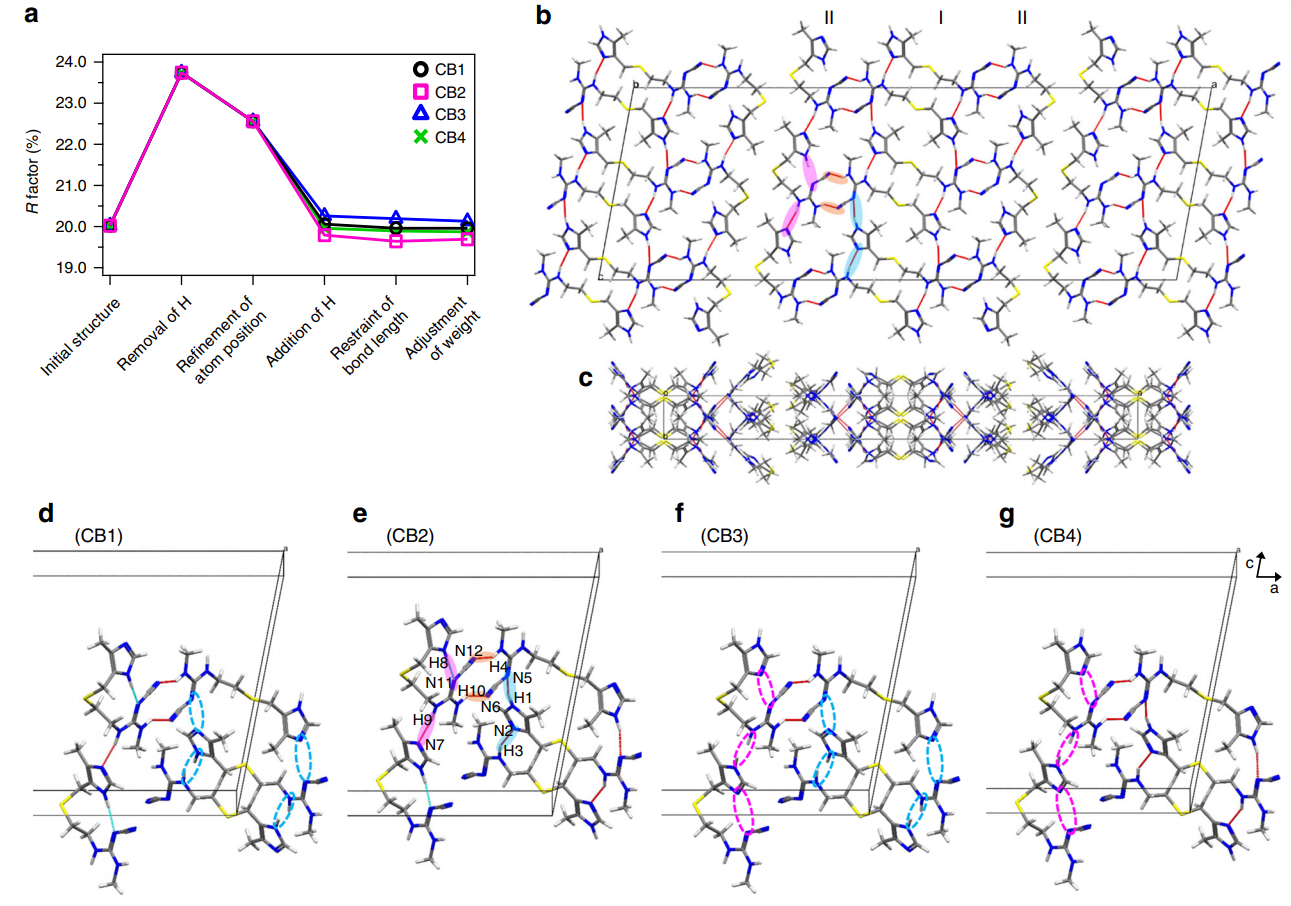
Figure 2: Cimetidine form B (CB) crystal structure and its hydrogen-bonding network. (a) Variation of the R factor (%) after each refinement step as determined using the SHELXL software. Hydrogen-bonding network of the CB2 structure in (b) ac and (c) ab planes. For the sake of simplicity, only intermolecular hydrogen bonds are shown as red lines. (b), (e) The pink and sky-blue solid circles indicate the hydrogen bonds between I conformations and between II conformations, respectively. The orange solid circles represents the hydrogen bonds between I and II conformations. (d)–(g) Extraction of hydrogen bonds for the (d) CB1, (e) CB2, (f) CB3, and (g) CB4 structures. The unit cell consists of adjacent molecules connected by intermolecular hydrogen bonds. The inter- and intramolecular hydrogen bonds are depicted by red and sky-blue lines, respectively. The magenta and sky-blue broken circles denote the missing hydrogen bonds of H8···N11 and H9···N7, and H1···N5 and H3···N2, respectively. Reproduced in accordance with Creative Commons Attribution 4.0 International License from reference [1].
NMR crystallography in the future
The crystalline structure of a compound, including its hydrogen atoms, can now be analyzed from nano- to micro-crystals of 100 nm to 1 µm, from mixed samples, using an ED and NMR crystallography approach.
Analytical techniques such as NMR crystallography will not only contribute to more stable quality assurance of low-molecular-weight pharmaceutical tablets and powders in the future, but could be leveraged to discover other unknown crystal forms that traditional diffraction techniques have so far failed to illuminate. Applications may extend beyond pharmaceuticals, to characterize various microcrystalline structural and functional chemical materials, such as porous metal complexes and metal-organic frameworks.
References
- Guzmán-Afonso C, Hong Y, Colaux H, Iijima H, Saitow A, Fukumura T, Aoyama Y, Motoki S, Oikawa T, Yamazaki T, Yonekura K and Nishiyama Y. Understanding hydrogen-binding structures of molecular crystals via electron and NMR crystallography, Nature communications, 2019;10: 3537.
- Hübschle CB, Sheldrick GM, Dittrich B. ShelXle: a Qt graphical user interface for SHELXL, J Appl Crystallogr, 2011;44(Pt 6):1281–1284.
About the Author(s):
Dr. Michael Frey
Michael receive his chemistry degree from Temple University and PhD in Biological Solid-State NMR from the University of Pennsylvania. He was a post-doc at the ETH in Zürich working on Inverse Detection Bio-NMR. Michael joined JEOL USA in 1984 starting in NMR applications and has worked in a variety of NMR areas including software development and NMR spectrometer R&D. He is currently the Analytical Instruments Product Manager and is responsible for NMR, ESR, and Mass Spectrometry.
Michael can be reached at frey@jeol.com.
Dr. Yusuke Nishiyama
Yusuke received his PhD from Kyoto University 2002. He was a post-doc at RIKEN working on manipulation of nuclear spin in solid-state NMR. Yusuke joined JEOL in 2007 as an application chemist especially for solid-state NMR. He established his research lab at RIKEN-JEOL collaboration center in 2014 and is working on interdisciplinary chemistry including NMR, TEM and computation. He is currently the research scientist at JEOL RESONANCE Inc., unit leader of nanocrystallography unit of RIKEN-JEOL Collaboration center, and deputy group director of NMR research and collaboration group, NMR science and development division at RIKEN SPring-8 center.
About JEOL
JEOL is a world leader in providing instrumentation for high industrial research and development. Core product groups include analytical instruments such as mass spectrometers, NMRs and ESRs, electron microscopes (SEMs and TEMs), instruments for the semiconductor industry (electron beam lithography and a series of defect review and inspection tools).
JEOL USA, Inc., is a wholly owned subsidiary of JEOL, Ltd., Japan, was incorporated in the United States in 1962. The company has 13 regional service centers that offer unlimited emergency service and support in the U.S.
For more information about JEOL USA, Inc. or any JEOL products, visit www.jeolusa.com, or call 978-535-5900.